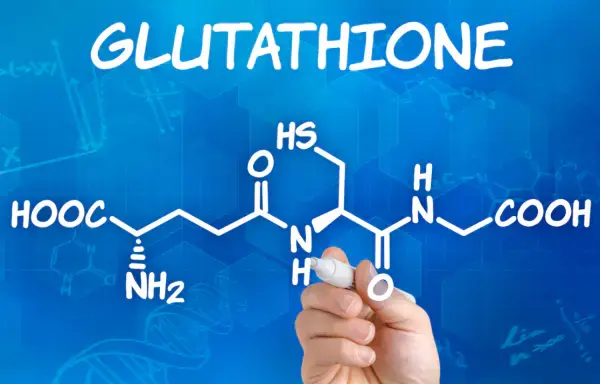
glutathione (GSH) is a tripeptide composed of glutamic acid, cysteine and glycine, containing sulfhydryl, which has antioxidant and integrated detoxification effects.
The thiol group on cysteine is the active group of glutathione (so glutathione is often abbreviated as G-SH), which is easy to combine with some drugs (such as paracetamol) and toxins (such as free radicals, iodoacetic acid, mustard gas, lead, mercury, arsenic and other heavy metals), and has an integrated detoxification effect.
Therefore, glutathione (especially glutathione in liver cells) can participate in biological transformation, thereby transforming harmful poisons in the body into harmless substances and excreting out of the body.

Glutathione also helps maintain normal immune system function.
Related literature
【 Title 】 : Blocking glycine utilization inhibits multiple myeloma progression by disrupting glutathione balance
【 Key words 】 Multiple myeloma
【 Literature content 】
Metabolites in the tumor microenvironment are key factors in tumor progression.
The lack of understanding of metabolic characteristics in the bone marrow (BM) microenvironment in multiple myeloma (MM) has limited our understanding of the progression of MM.
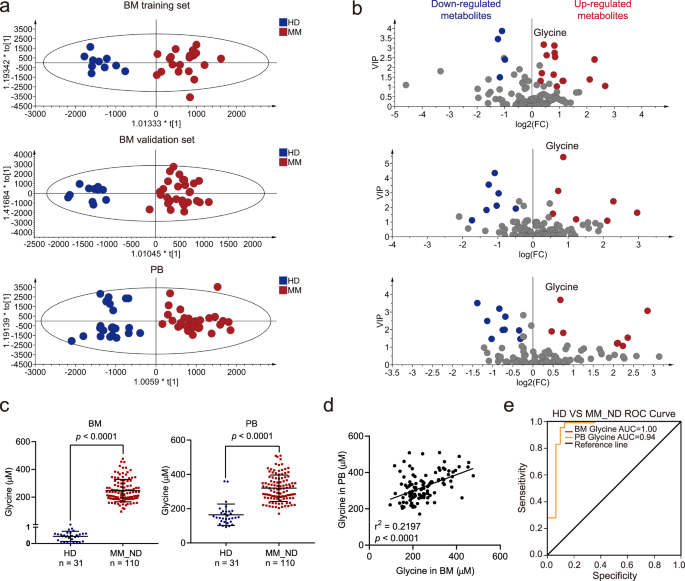
Here, we show that glycine concentration is elevated in the BM microenvironment due to collagen degradation mediated by matrix metallopeptidase 13 (MMP13) secreted by MM cells, and that elevated glycine levels are associated with MM progression.
MM cells use channel protein solute carrier family 6 member 9 (SLC6A9) to absorb exogenous glycine and subsequently participate in glutathione (GSH) and purine synthesis.
Inhibition of glycine utilization by SLC6A9 knockdown or betaine treatment inhibited MM cell proliferation and enhanced the effect of bortezomib on MM cells.
We found that glycine is a key metabolic regulator of MM, revealing molecular mechanisms that control MM progression and providing promising therapeutic strategies for MM therapy.
【 Title 】 : Phenylpropionic acid produced by gut microbiota alleviates acetaminophen-induced hepatotoxicity
【 Key words 】 Liver injury
【 Literature content 】
Gut microbiota affects liver drug metabolism. However, the gut microbiome factors that regulate drug metabolism in the liver are largely unknown.
In this study, using a mouse model of acetaminophen (APAP) -induced hepatotoxicity, we identified an intestinal bacterial metabolite that controls liver CYP2E1 expression and thus catalyzes the conversion of APAP into an active toxic metabolite.
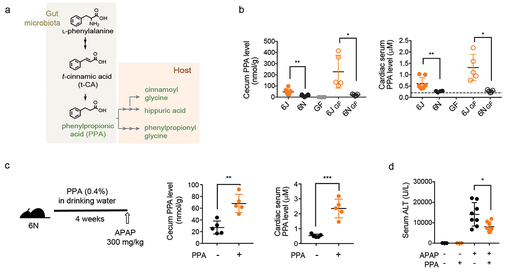
By comparing C57BL/6 subline mice from two different suppliers, Jackson (6J) and Taconic (6N), which are genetically similar but have different gut microbiomes, we determined that differences in gut microbiomes lead to different sensitivities to APAP-induced hepatotoxicity.
6J mice less sensitive to APAP-induced hepatotoxicity than 6N mice, and this phenotypic difference reproduced in germ-free mice by microbiota transplantation.
Phenylpropionic acid (PPA) identified by comparative non-targeted metabolomics analysis of portal vein serum and liver tissue from conventional and regularized 6J and 6N mice, which had higher levels of PPA.
PPA supplementation mitigated APAP-induced liver toxicity in 6N mice by reducing liver CYP2E1 levels. In addition, PPA supplementation mitigated liver damage induced by CYP2E1-mediated carbon tetrachloride.
Our data suggest that a previously known PPA biosynthetic pathway is responsible for PPA production.
Surprisingly, while PPA in the cecal contents of 6N mice almost undetectable, the 6N cecal microbiota produced PPA in vitro,
just as the 6J cecal microbiota did, suggesting that PPA production in the 6N gut microbiota suppressed in vivo.
No previously known gut bacteria with PPA biosynthetic pathways detected in either the 6J or 6N microbiota,
suggesting the presence of as-yet-unidentified PPA-producing gut microbes.
Our study reveals a novel biological function of the gut bacterial metabolite PPA in the enterohepatic axis and lays an important foundation for the study of PPA as a regulator of CYP2E1-mediated liver injury and metabolic disease.
【 Key words and products 】 Autophagy, oxidative stress
【 Literature content 】
Autophagy primarily activated by cellular stress, such as starvation or mitochondrial damage.
In several cell types, such as thymus epithelial cells (TEC), stress-independent autophagy activated by an unknown mechanism.
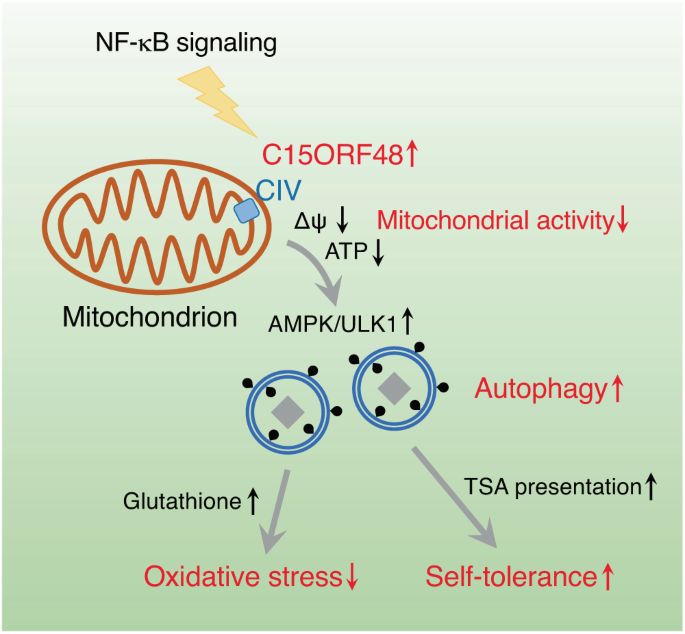
We report here that mitochondrial protein C15ORF48 is a key inducer of stress-independent autophagy.
Mechanistically, C15ORF48 reduces mitochondrial membrane potential and reduces intracellular ATP levels,
thereby activating AMP-activated protein kinase and its downstream UNC-51-like kinase.
Interestingly, C15ORF48-dependent autophagy induced upregulation of intracellular glutathione levels, promoting cell survival by reducing oxidative stress.
Mice lacking C15orf48 showed a stress-independent reduction in autophagy in TEC,
but no reduction in the typical hunger-induced autophagy in skeletal muscle.
C15orf48 — / — mice develop autoimmunity,
which is consistent with the fact that non-stress autophagy in TEC is essential for thymus autotolerance.
These results suggest that C15ORF48 induces non-stress autophagy, thereby regulating oxidative stress and autotolerance.
【 Key words and products 】 Asbestos fiber, oxidative stress
【 Literature content 】
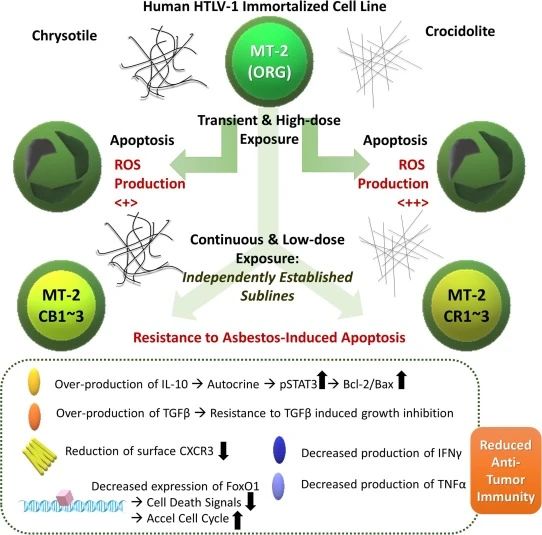
The effects of asbestos fibers on human immune cells have not fully established. We developed a cell line model of sustained exposure using the human T lymphocytic virus 1 (HTLV-1) immortalized human T cell line MT-2.
Sublines continuously exposed to chrysotile asbestos (CH) or chrysotile asbestos (CR) exhibit acquired resistance to asbestos-induced apoptosis after brief and high dose re-exposure to the fiber.
These sublines, as well as other immune cells exposed to asbestos,
such as natural killer cells or cytotoxic T lymphocytes, showed decreased anti-tumor immunity.
In this study, the expression of genes and molecules associated with antioxidant stress examined.
Complexes associated with oxidative phosphorylation have been studied because the production of reactive oxygen species (ROS) is important when considering the role of asbestos in carcinogenesis and the mechanism of resistance to asbestos-induced apoptosis.
In sublines with continuous exposure to CH or CR, thioredoxin expression reduced.
Interestingly, the expression of nicotinamide nucleotide transhydrogenase (NNT) significantly enhanced. Therefore, NNT knockdowns subsequently performed.
Although knockdown clones did not show any changes in proliferation or apoptosis,
these clones showed a recovery in ROS production and a recovery in the NADPH/NADP+ ratio,
whereas a reduction in ROS production in the persistently exposed sublines led to an increase in the NADPH/NADP+ ratio.
These results suggest that NNT is a key factor in preventing ROS-induced cytotoxicity in T cells continuously exposed to asbestos.
Given that these sublines exhibit reduced anti-tumor immunity, modifying NNT may help restore the anti-tumor effects of asbestos-exposed T cells.
【 Literature content 】
The dietary conditions of the father may lead to metabolic disorders in the offspring.
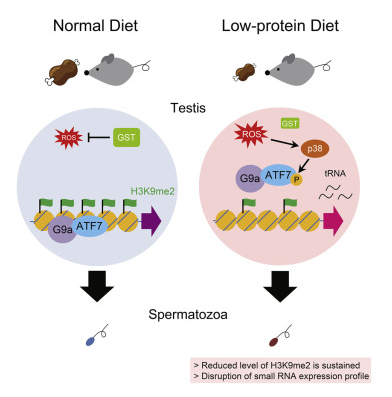
We analyzed the role of the stress-dependent epigenetic regulator cyclic adenosine-dependent transcription factor 7 (ATF7) in liver gene expression changes induced by a paternal low-protein diet (pLPD) in mice.
The Atf7 +/ – mutation resulted in offspring with a phenotype similar to that caused by pLPD,
and the effects of pLPD almost disappeared when paternal Atf7 +/ – mice used.
ATF7 binds to the promoter region of about 2,300 genes in testicular germ cells (TGC),
including genes associated with cholesterol biosynthesis and tRNA genes.
LPD induces the phosphorylation of ATF7 by p38 through reactive oxygen species (ROS) in TGC. This results in the release of ATF7 and reduced dimethylation of the histone H3K9 (H3K9me2) on its target gene.
These epigenetic changes maintained and induce the expression of certain tRNA fragments in sperm. These results suggest that LPD-induced and ATF7-dependent TGC epigenetic changes play an important role in paternal diet-induced metabolic reprogramming of offspring.